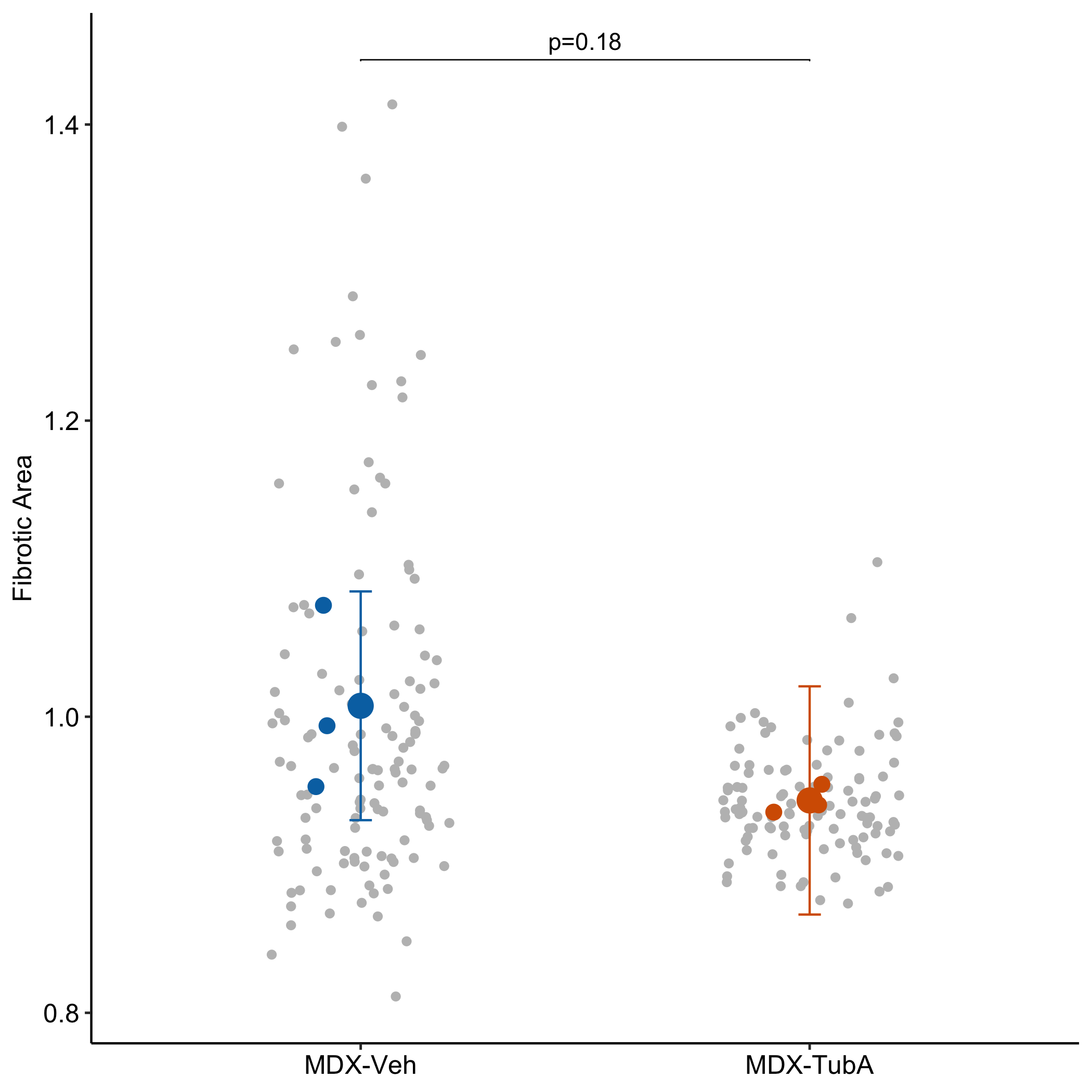
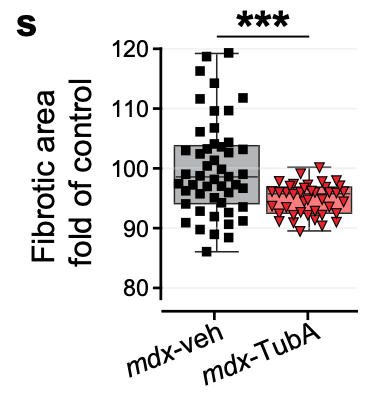
Fig 2s – Pharmacological inhibition of HDAC6 improves muscle phenotypes in dystrophin-deficient mice by downregulating TGF-β via Smad3 acetylation
Fig 2s is a Completely Randomized Design with subsampling (CRDS), with 28-49 technical replicates per mouse. The researchers used a Mann-Whitney test that ignored the technical replication. This analysis is massively pseudoreplicated because techanical replicates are not independent evidence of effect of treatment. Individual mouse ID was archived, which makes it easy to re-analyze with models that account for the non-independence. The p-value accounting for non-independence is 0.18 while the p-value using Mann-Whitney on all data is 0.0002. Whoops!
linear mixed model
random intercept
CRDS
nested ANOVA
ggplot
Author
Jeff Walker
Published
June 2, 2024
Modified
October 2, 2024
Fig 2s repro generated using ggplot. The technical replicates are de-emphasized by using a gray color.
Design: Completely Randomized Design with Subsampling (CRDS)
Response: fibrotic area
Key learning concepts: linear mixed model, random intercept, nested ANOVA
Quick learning explanation: Nested data are measures within a discrete unit or a hierarchy of units, for example technical replicates within a tumor within a mouse.Pseudoreplication is the analysis of subsampled (technical) replicates as if they were independent measures of treatment effects. They aren’t, because technical replicates within a unit (a mouse, a culture) share causes of variation that not shared by technical replicates in other units. This is a violation of the assumption of independence of errors.
Wilcoxon rank sum test with continuity correction
data: fig2s[treatment == treatment_levels[1], fibrotic_area] and fig2s[treatment == treatment_levels[2], fibrotic_area]
W = 8740, p-value = 0.000204
alternative hypothesis: true location shift is not equal to 0
A nested ANOVA is a classical method that is equivalent to the linear mixed model above when there are an equal number of technical replicates per mouse, but will be close otherwise. In general, things will rarely go wrong if one uses a nested ANOVA instead of the linear mixed-model. GraphPad Prism does nested ANOVA but not a linear mixed model.
The technical replicates are de-emphasized by using a gray color. The modeled means of each mouse are colored by treatment (these are the predicted means of the linear mixed model and not the simple mean of each mouse). The model means of each treatment are colord by treatment.